运用理论化学计算方法,进行光催化和太阳能电池材料结构与性能,以及光转化动力学的研究,是太阳能研究领域的重要方面,正在发挥着越来越大的作用。我组的研究方向主要集中在一下方面:(1)光电材料吸光性质的研究;(2)光电催化反应机理的模拟;(3)电子-空穴分离与传输的微观机制
TiO2作为光催化材料受到广泛的关注,但由于其带隙太宽,对太阳光中的可见光部分吸收很少,直接影响其作为光催化材料的光利用效率。我们通过第一性原理计算,对(M+N)共掺杂元素 (M代表Nb,Ta,Mo,W等金属元素;N代表C,N等非金属元素) 在TiO2中的溶解性以及掺杂后对TiO2能带结构的影响进行了研究,研究发现:(i)在富氧条件下,(W + C)和(Ta + N)在TiO2中有很好的溶解性,掺杂原子主要进入Ti和O原子的晶格位,这样可以有效减少复合中心的形成;(ii) (Ta + N) 和 (W + C)共掺杂TiO2的带隙宽度明显减少,导带边的位置几乎没有变化,说明掺杂可以提高TiO2的对可见光的吸收效率,同时未影响TiO2的还原能力。另外我们的计算结果还从理论上解释了:(1) 实验中制备的(Mo+ C)共掺杂TiO2的光催化性能远小于文献中报道的理论结果;(2)实验表明(W+N)共掺杂TiO2的催化效率明显高于(Mo+N)共掺杂的TiO2。
我们利用密度泛函方法研究了具有固溶体结构的光催化材料ZnxCd1-xS的结构与性质。我们通过SQS方法建立了不同原子比例的固溶体模型(ZnxCd1-xS),并通过第一性原理计算(使用的是HSE06泛函)得到了固溶体的带隙宽度随着原子比例(x)的变化。下图分别表示晶胞参数a/c以及带隙宽度随着x的变化趋势。图中可以看出 (i)晶胞参数随着x的增加呈线性减小的趋势;固溶体的带隙宽度随着x的增加而增加,增加的趋势可以用二次函数来拟合。这说明ZnxCd1-xS固溶体的物理特性可以用Vegard’s定律来很好的描述。(ii)我们计算得到的结果和实验值符合的很好,这说明通过我们的计算模型和方法可以对固溶体体系的性质做出很好的预测。
此外,我们开发了一种可以计算纳米半导体材料这种超大体系被紫外和可见光激发后的结构与性质的理论计算方法和软件。该软件包软件包括了在Linux环境下用fortran90编写的Fortran计算部分,和windows下面的用visual basic编写的界面部分。其特点为将紧束缚和密度泛函方法结合,采用一些适用于大体系的算法,使得计算速度接近半经验的紧束缚方法,而计算精度接近于从头算的密度泛函方法。从而非常适用于大体系的电子结构和性质的计算。同时拥有windows下面的可视化界面,从而使得计算起来非常简单。程序的特点归纳为:使用简单,快速高效。在计算速度方面,与其它方法相比,对于相同的体系计算时间有了大幅度的缩短,计算所占用的CPU时间比INDO/S等半经验方法还要快十几倍,为计算大分子激发态提供了必要的前提。而关于计算精度方面TD-DFTB方法与其它高精度算法相比,也达到了可观的计算精度,误差范围控制在10%之内。可见对于大分子体系,其计算的优势是明显的。
在完成BDFT程序包的编写工作后,我们用该程序研究了光电材料ZnO纳米团簇的结构和光谱性质。对于低维结构的研究,前人对提出了ZnO小分子团簇的环状、笼状等结构,对于原子数目较多的ZnO大分子团簇,一直没有得到合理的结构。通过DFTB方法的计算,以截取六方纤锌体晶体结构作为初始构型,进行所有原子的全优化。我们得到了大分子团簇的稳定结构,其主要以笼状结构为主,并且Zn原子向团簇中心移动,而O原子向外移动。这对于表面原子的钝化和修饰是很有指导意义的,说明钝化剂和修饰物一般先与O原子成键。
对于大分子团簇,我们第一次给出了低激发态的计算结果,有力的验证了量子限制效应。一般而言,量子限制效应在材料的尺度小于或等于波尔激子半径时候产生,然而对于ZnO纳米粒子,在尺度大于波尔激子半径2~3倍的时候,量子限制效应就已明显产生。对于表面效应,我们选取了两种团簇模型,一种是表面含有孤立原子的,一种是表面平滑的。发现其低激发态的阈值有很大的不同,随着模型尺度的变化,这种差异并未消失。对于纳米粒子处于有机酸的环境中,我们计算了其红外谱,结果与实验较好的符合。
开发能响应可见光,同时具有高量子产率的光催化剂一直是光催化材料研究的重要方向。处在d 区具有d0 电子构型的化合物如TiO2, SrTiO3 , K4Nb6O17 , 以及处在p区具有d10电子构型的MIn2O4 (M= Ca ,Sr) , Sr2SnO4 , NaSbO3和Zn2GeO4等被广泛研究。但是这些催化剂只能响应占太阳光谱约4 %的紫外光,对太阳能的利用率很低。
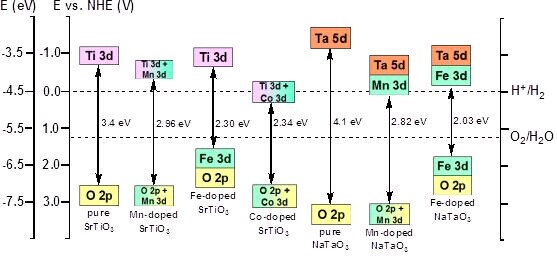
利用第一原理计算,运用离子掺杂的方法,从微观上调变材料的能带结构。首先选取了在紫外光区具有很高光催化活性的SrTiO3和NaTaO3作为研究对象。研究结果发现,在这两种晶体中,掺杂的金属Mn, Fe, Co更易于取代离子半径相近的Ti和Ta。实验中观测到金属掺杂的SrTiO3具有可见光吸收的原因是掺杂金属的3d轨道对材料的价带顶和导带底有重要贡献,使材料的能带明显减小。
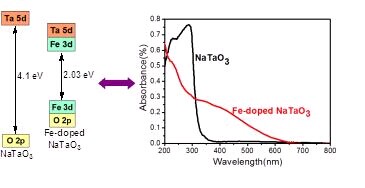
Xin Zhou, Jingying Shi, Can Li, “Effect of Metal Doping on Electronic Structure and Visible Light Absorption of SrTiO3 and NaTaO3 (Metal=Mn, Fe, and Co)”, J. Phys. Chem. C, 2011, 115, 8305



